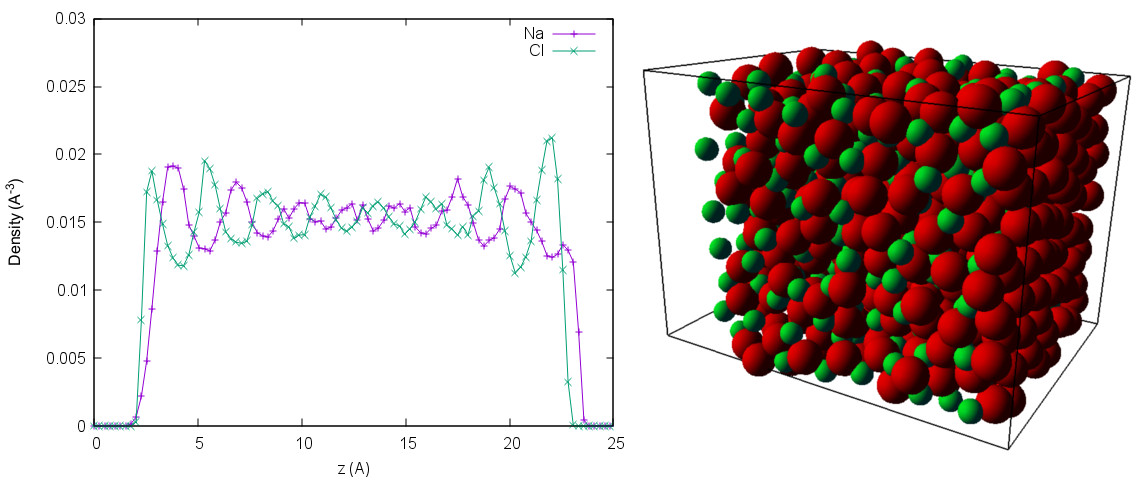
['ADDITIONAL_CHECKS', 'AFFINITY', 'BMHTF_NACL', 'BOND_CONSTRAINT', 'BUCKINGHAM', 'COLLISION_DETECTION', 'CUDA', 'DIPOLAR_BARNES_HUT', 'DIPOLAR_DIRECT_SUM', 'DIPOLES', 'DP3M', 'DPD', 'EK_BOUNDARIES', 'ELECTROKINETICS', 'ELECTROSTATICS', 'ENGINE', 'EXCLUSIONS', 'EXPERIMENTAL_FEATURES', 'EXTERNAL_FORCES', 'FFTW', 'GAUSSIAN', 'GAY_BERNE', 'GSL', 'H5MD', 'HAT', 'HERTZIAN', 'LANGEVIN_PER_PARTICLE', 'LB_BOUNDARIES', 'LB_BOUNDARIES_GPU', 'LB_ELECTROHYDRODYNAMICS', 'LENNARD_JONES', 'LENNARD_JONES_GENERIC', 'LJCOS', 'LJCOS2', 'LJGEN_SOFTCORE', 'MASS', 'MEMBRANE_COLLISION', 'METADYNAMICS', 'MMM1D_GPU', 'MORSE', 'NPT', 'P3M', 'PARTICLE_ANISOTROPY', 'ROTATION', 'ROTATIONAL_INERTIA', 'SMOOTH_STEP', 'SOFT_SPHERE', 'TABULATED', 'THOLE', 'VIRTUAL_SITES', 'VIRTUAL_SITES_INERTIALESS_TRACERS', 'VIRTUAL_SITES_RELATIVE', 'WCA']